|
|
|
 |
Selected
Talks
ASN Board Review Course, San Francisco. August, 2005 Case Discussions: Disorders of Calcium Metabolism |
| |||||||||||||||||||||||||||||||||||||||||||||||||||
00:01

Introduction and Case 1.
For the next hour and a half, we have a wonderful session to kind of bring together all of the concepts that we discussed this morning. I believe Dr. Bushinsky is going to start with the first case, and I will just hand over the podium to him.
Dr. Bushinsky: Be cognizant as you're eating your lunch of the phosphate load that you are now ingesting and telling your patients, of course, to avoid. That is the first introduction to this first case. We each have two cases. We'll just go in a round robin. This is made to be interactive. So speak up. If you have a question, do not wait for me to ask for your questions. Speak up.
First patient: These are real patients; at least this is a real patient that I saw a number of years ago. A 35-year-old white male with adult-onset PKD. PKD was diagnosed at age 27 while he was in the Army. He was discharged for medical reasons. No follow up. He was adopted, so unfortunately we have no family history, and he had a limited education and had a very poor understanding of this disease.
01:33

Case 1: Labs
He had back pain and polyuria. We did an ultrasound. The right kidney was large. The left kidney was larger. There were numerous cysts. The bicarbonate concentration was 15, low, suggesting that he could not excrete his endogenous acids. His creatinine was 9.3, BUN 111. Calcium was 11. Phosphorus was 9.1, clearly elevated. His alkaline phosphatase was also up. We talked to the patient about phosphate binders. Unfortunately, he delayed getting a vascular access.
02:19
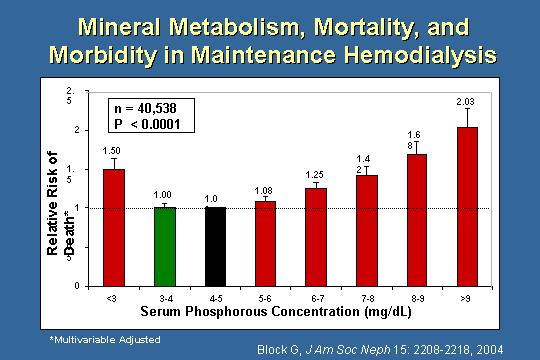
Serum phosphorus concentration
And let's think about the calcium and phosphorus in this gentleman. You saw the slide already this morning, and I bring it up again because Geoff Block has a study from 1998. This is a similar study in 2004. Others have similar data. Multivariate-adjusted. It is clear that an increase in serum phosphorus concentration is associated with an increase in a relative risk of death. A marked decrease in phosphorus is also associated with a significant increase in the relative risk of death. It is not a perfect study, as Miles said to us, we do not know about vitamin D, we do not know about a lot of other things; but clearly, would I rather have a phosphorus of 4 to 5 or one that is greater than 9.0? It is clear which group I would rather be in.
03:24
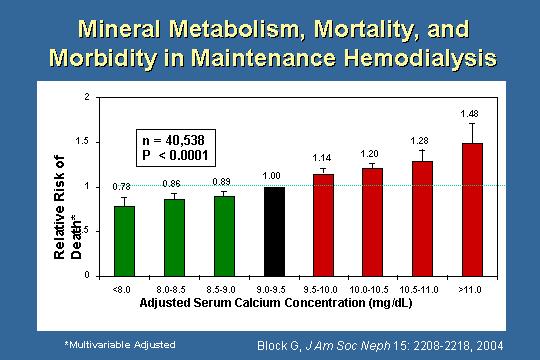
Adjusted serum calcium concentration
We also know that with the adjusted serum calcium there is also a significant correlation; higher calciums increase the risk of death. Lower calciums looks like they decreases the risk of death. So, the hypocalcemia that I worry about my dialysis patients, I do not worry about quite as much anymore. What I show these two slides for is for the following.
03:58
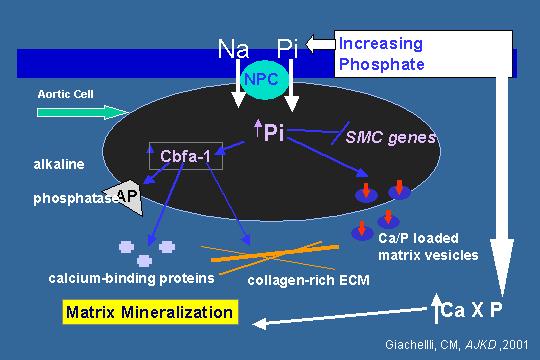
Matrix mineralization
There is very nice data now. This is a 2001 cartoon, but if you look at the Current Opinion in Nephrology and Hypertension Shanahan has similar data to Giachelli on what happens to vascular smooth muscle cells. This is an aortic cell. As you increase phosphate, you increase the number of sodium phosphate co-transporters. I have been calling it NaPi, you can call NPC, which increases the intracellular phosphate. You have upregulation of several vascular smooth muscle cell genes: Cbfa-1 goes up and this gene is clearly associated with mineralization, not only in vascular smooth muscle cells and bone cells, but in renal tubular cells that are calcifying as well. You have elevated alkaline phosphatase, elevated calcium-binding proteins, elevated collagen-rich extracellular matrix, and it is thought by some that you have these calcium-phosphate loaded matrix vesicles that move calcium into the collagen-rich extracellular matrix and mineralize it, and that the increased calcium and phosphorus in the blood leads to increased matrix mineralization.
We are calcifying our dialysis patients. As you know, the risk of death in a 30-year-old dialysis patient is equal to that of a 75-year-old to 80-year-old nondialysis patient by Foley's data, whether the patient is a black or white male or female, and much of this is cardiovascular mortality, and we were starting to understand how an elevation in phosphorus and an elevation in calcium might increase cardiovascular mortality. End of lecture. Back to our case.
06:11

+3 months began hemodialysis
Three months later, the patient began on hemodialysis. Creatinine was 9.1, BUN was 134. It took this guy 3 months to get started on dialysis. Calcium remained elevated. Phosphorus also remained elevated. Many missed treatments; many abbreviated treatments. He was diagnosed as ADHD. We have heard about ADH with water transport. We heard about ADH with autosomal dominant hypocalciuria or hypercalcemia. We now talk about ADHD, which is attention deficit hyperactivity disorder. He had borderline intelligence on formal testing.
06:55

+4 months - admitted
He was admitted to the hospital 4 months later with an infected renal cyst; he had Klebsiella. Calcium was elevated, phosphorus was elevated. His PTH was now over a 1000 pg/mL. His 1,25 D level was low. His 25,D level, which unfortunately we were not smart enough to measure at that time, was also low. We did a parathyroid ultrasound and you can see the right upper pole, he had a big parathyroid. The left upper pole also shows a big parathyroid. We saw one gland with the ultrasound on each side. On chest x-ray he had extensive soft tissue calcification. We could not use cinacalcet on him because it was before the cinacalcet era, and he refused parathyroidectomy. He was clearly judged to be competent.
07:49
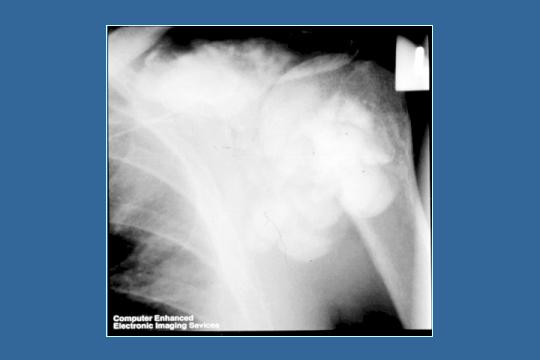
Right shoulder x-ray
I do not know how well you can see it, but this is pure calcification of his shoulder due to the extensive calcium phosphate deposition. When you raise the supersaturation through the mechanisms we have shown, you get calcification.
08:06

+8 months
He refused all meds. He intermittently showed up for dialysis. He finally agreed to a parathyroidectomy. He had calciphylaxis, ulcerations, multiple soft tissue lesions.
08:19
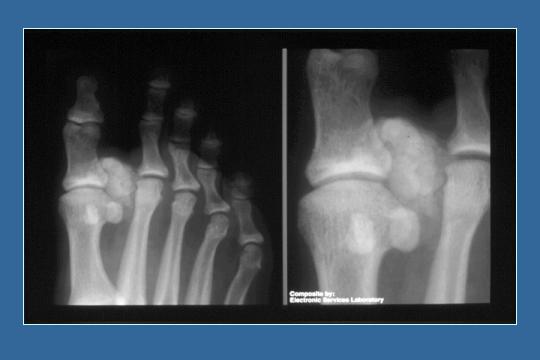
Calcifications - x-ray of foot
This just shows more calcification.
08:23
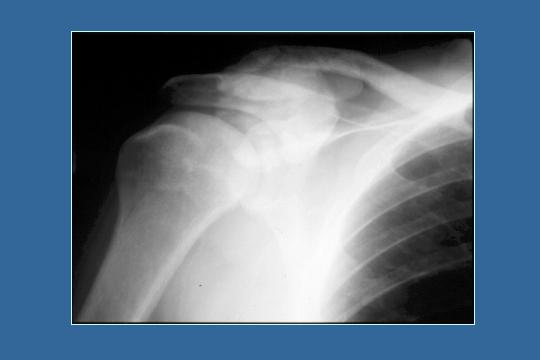
Calcification - x-ray right shoulder
...more calcification.
08:27

Surgery
At surgery, we found two glands, 1 cm and 2 cm, on the left side -- these are big glands. The right upper gland was 1.5 cm. The right lower gland could not be identified. Our surgeon looked at the entire lobe of his thyroid. He resected right lower lobe but could not find it. He reimplanted parathyroid tissue into the right forearm. You can judge whether he should have reimplanted tissue or if he should have left one of these three big glands. That is personal preference. As long as you have a good surgeon, it does not matter what they do. Most surgeons are leaving some tissue behind. At day 6, the the serum calcium had fallen a little, then started to rise. The serum PTH was 972.
What is going on? Don't all speak at once! I cannot hear you. So, what do you think is going on? I cannot hear you, but I assume that all of you are thinking he has an accessory parathyroid gland. A small percentage, but not that small a number of people have the fourth gland in the mediastinum, even in the abdomen, and you have to look for it, because clearly his PTH level after surgery should be trivial (very low) , but it is still 1000. So we went looking.
09:59

Investigation for ectopic parathyroid gland
We did an MR of the neck. No evidence of the parathyroid gland; and then we did a subtraction scan. I am going to show you in this case and another case why the sestamibi scan is better than the subtraction scan, and if your nuclear medicine staff is not doing it, then you should convince them to do it. Intense activity in the inferior portion of the thyroid and to the right of the manubriosternal joint. He continued to be noncompliant. Our psychiatry friends said that he was competent.
10:34
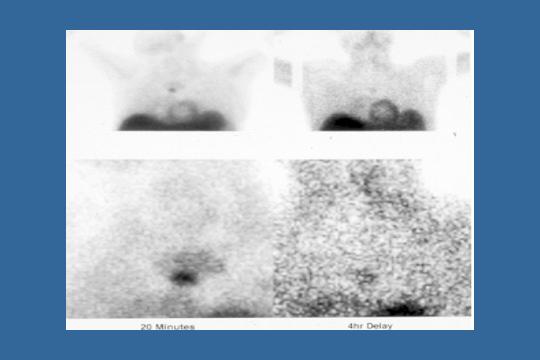
Sestamibi - parathyroid
This is, I guess, the sestamibi scan. You can see a parathyroid gland lighting up right there. You have to go after that and you have to get that gland. Otherwise, the PTH will never, never fall.
10:53

Calciphylaxis
Our patient would not let us do it. He would drink large amounts of milk brought by his family. He would take milk off other patient's trays. A quart of milk is the best phosphate replacement. If you want oral phosphate replacement, there is 1 gm of phosphorus in a quart of milk. Give your (hyophosphatemic) patients milk to drink. The hypophosphatemia causing a respiratory paralysis that Miles told you about is also seen during refeeding using any other type of food, and the best therapy is simply milk. It is cheap. It is good. The patient made himself DNR; Serum calcium - 11, serum phosphorus - 11, and the PTH was now up to 1300 from that single gland. He became septic and hypotensive and died.
11:45

Postmortem
They found a fourth parathyroid gland postmortem exactly where the scan showed and it was a big gland. A woman during the break told me about a very similar case in which they found three glands but they could not find the fourth, and the surgeon went looking for it in the mediastinum and could not find it. It is there. You just have to get the right surgeon.
12:09
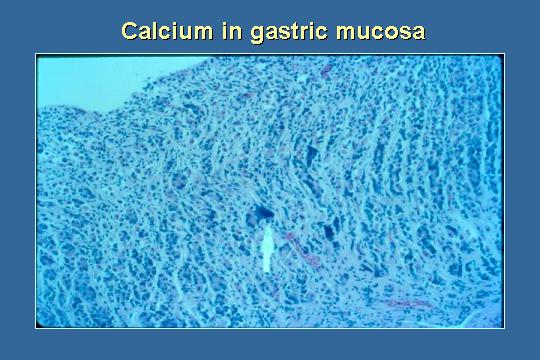
Calcification
If you look at the gastric mucosa, we see some calcification. Why should there be calcification in the gastric mucosa? Think about the physiology of the gastric mucosa. Why do you often see calcification there? Someone said pH, I think, or maybe they said phosphate. As you secrete hydrogen ions into the lumen, you put out bicarbonate on the basolateral side. That bicarbonate will raise the pH. We know calcium phosphate is not as soluble in an alkaline milieu, and the patient calcifies the gastric mucosa.
13:00
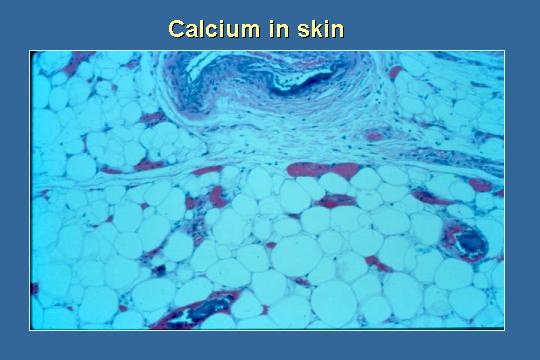
Calcium in skin
There was calcium in the skin. This is clearly calciphylaxis.
13:03
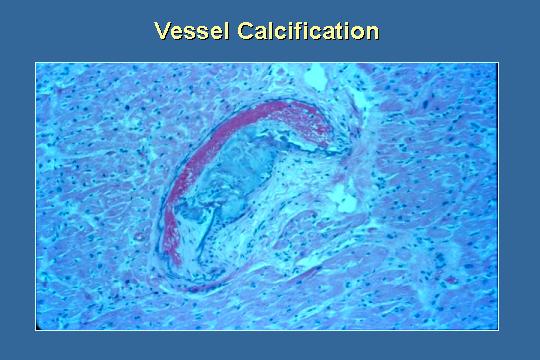
Vessel calcification
Here is a vessel with some calcification...
13:09
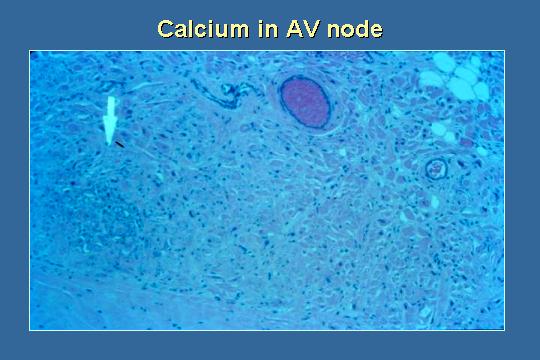
Calcium in AV node
...and there was calcification in the AV node even, and this patient died because of his high calcium and phosphorus, probably phosphorus more than calcium, and it is just to show you the ravages of high calcium and phosphorus and also of hyperparathyroidism.
13:27

Questions and Comments
Audience: Should the surgeon have checked intraop PTH levels?
Dr. Bushinsky: That is a nice trick, but they could not find the fourth gland. Yes, absolutely.
Audience: Before you implant more parathyroid tissue, should you make sure you have no parathyroid secretion?
Dr. Bushinsky: PTH has a half-life of minutes. You are going to see a very rapid fall in serum calcium and PTH. It might have made sense before you do the implantation to make sure the PTH level fell. You can always freeze the parathyroid tissue and implant it later; this works very well. Other questions?
Audience: Should you or should you not do ultrasound of the parathyroid glands?
Dr. Bushinsky: If you are going to be cost-effective, you should not do it. There is really in this case little reason to have done it. At an academic institution, if the resident orders it, I tend not to say, "Do not do it", but you are absolutely right. There is really no reason in a case like this to go and do a preop ultrasound. There is no reason to do preop sestamibi scans, except if you are worried about that FHH that we have talked about so much this morning.
Audience: There are a few case reports regarding the role of bisphosphonates in calciphylaxis, and also there are several case reports of the role of bisphosphonates in extravascular calcification, I am just wondering, is there any role for giving bisphosphonates in this case?
Dr. Bushinsky: It is clear that calciphylaxis can be associated with a high and a low PTH. So, in my mind, there is no role for parathyroidectomy, not in this case, but in the typical case of calciphylaxis. Is there a role of bisphosphates? As far as I know, there has never been a controlled study. All of it is, as you say, in case reports. I could understand how it could work, but there is no data. So, I do not advocate it.
Audience: I guess there are several case series in extravascular calcifications like infantile vascular calcification that showed that bisphosphonates are useful, and there are a few studies that showed it controls the refractory hyperparathyroidism, and there are several case series other than vascular calcification, where in a dystrophic calcification use of bisphosphonates is also helpful. I guess it could be used. In this case, we did not have any option I guess?
Dr. Bushinsky: We did not. Until there is a real controlled study, we have to understand that, with any case series, people publish what works. They do not publish what does not work. So, what you see in the literature is a best-case estimate of what possibly could happen. Until there is a controlled double-blind study, all bets are off.
Audience: There are some studies about use of sodium thiosulfate in calciphylaxis. Can this be used also in the soft tissue calcifications?
Dr. Bushinsky: Again, it is the same issue. The use of thiosulfate makes sense. It inhibits crystallization in the urine, but as far as I know, there is no clear data. Do you think it works or not?
Unfortunate case in my mind. Hyperphosphatemia is the biggest risk to our dialysis patients. We control it with calcium-containing binders to the limit as recommended by KDOQI and a large use of noncalcium-containing phosphate binders.
18:28

Dr. Andress: Case 2
I have one slide, so you're going to have to help. A 47-year-old African American man who is a smoker. Referred by the primary care doctor to you for management of hypercalcemia. He has a past medical history of hypertension, for which he was treated with metoprolol. Three months earlier, he was started on hydrochlorothiazide because this came out in a journal as being life prolonging.
19:01

Case 2: Labs
Here are the lab tests that come with him in this referral: sodium of 139, K of 3.2, chloride of 103, and bicarb of 27. His renal function appears absolutely normal. Glucose is 104. The calcium is indeed high at 12.2. The phosphate is 4.4. You rifle through the records, and you find that the serum calcium has been undergoing a slow, steady rise over the last year and a half, a high normal 9.7, subsequently 9.9 a year ago, and six months ago it was still within the normal range, but again, at the high end. That is the case.
19:48

Case 2: Diagnosis
So what are the possibilities? What tests should be ordered and then what is the treatment? Let me go back and just leave that posted. I guess the first step is, where do we start? I think the place we start is what they sent the patient to us for, and what is the most outstanding lab value that is presented, and that is obviously the hypercalcemia. So, what do we need to do?
Could you just stop the thiazide? This is one possibility. I think what you might be proposing is one of the issues that comes out in this case - that the thiazide was responsible for his hypercalcemia kicking off in only the last few months, because before it was really somewhat stable, although slowly rising; but in the last three months, the serum calcium has risen to 12.2, and clearly the thiazide could be implicated as exacerbating his hypercalcemia.
Do you think that the thiazide is accounting for the whole problem? Possible, but not likely. Certainly, one thing would be to get rid of the thiazide. Somebody wanted to order a PTH. Anybody want to order an ionized calcium just to be sure? I think we are going to have one of those in our pocket. When you evaluate hyponatremia once, you just want to make sure that the osmolality is actually low too and then you go down the whole pathway. It is annoying to have to remember it after you have done million tests and not have an answer. Let say the ionized calcium here was on the high side. Let say it was 1.44. We are in fact talking about hypercalcemia.
Somebody wants to order a PTH, but before we get to ordering tests, what things are crossing your mind based on the information we know?
Audience: Cancer is one possibility.
Dr. Bushinsky: If it was cancer, what type of mechanism would you be proposing?
Audience: PTHrP-mediated.
Dr. Bushinsky: What are typical players for that? Squamous cell carcinoma of the lung. Certainly, he is a smoker and that is a possibility. Renal cell would be a good one. Breast cancer is pretty rare in a man, but it occasionally happens, but that will be the big one in women. So, that is good. Anything to argue against that? The phosphate level. What would you anticipate? It should be low, because if you have excessive unregulated activation of either PTH or PTHrP, one would anticipate a lowish serum phosphate, maybe the serum phosphate values are not going to be terribly low, but they certainly should be on the higher side, as they are in this case. Most people have a serum phosphate between 2.5 and 4.5, but that means there are plenty of people down around 2.7 and 3. So, this value is a little on the high side. Of course, the other thing is just to make sure is that the serum creatinine is 0.8, because if the serum creatinine was 1.7, you might not put as much weight in the serum phosphate not being low. That makes it more believable. That is one possibility, certainly a malignancy. We are going to need to get a chest x-ray, no doubt. Anything else?
Audience: Vitamin D intoxication was reported.
Dr. Bushinsky: Let us see - would that be a possibility? That would give you hypercalcemia, certainly. Could the relatively high serum phosphate be explained by that? You might expect a highish phosphate, but I could live with that, that is not too bad.
When you have vitamin D intoxication and hypercalcemia often with hyperphosphatemia, what is the renal function typically? It is usually not pristine. This has the makings of something chronic rather than an acute presentation with a milk alkali syndrome, but that is certainly a possibility. So, we could consider getting what tests to address that?
You measure what? 25,D somebody said. Yes, the 25,D is how you always assay the storage form of vitamin D. So, that would be on the list of possibilities.
What else, going back to cancer, as we left that quickly? Sarcoid somebody said. Let us come back to sarcoid. PTHrP we said is a possibility. Any other possible etiologies of malignancy?
Audience: Myeloma.
Dr. Bushinsky: So, could myeloma fit this? Certainly, a little young, 47 years old. Maybe, if he is HIV positive, then it would really be possible. Then you can get those myelomas down into the age 30s and 40s, but it is possible. The phosphate is consistent with that. The lack of renal dysfunction is fine. Of course, many cases of myeloma present with renal dysfunction, but they do not have to. The issue we brought up with the anion gap this morning is cute if you find it, but if it is not there, it does not rule out myeloma by any means. That is something worth considering. Almost anybody who sets foot in the nephrologist's office ends up with an SPEP and a UPEP, so we will get that too.
25:08

Sarcoid
What about sarcoid? Who is the sarcoid champion? What is your thought? Possible? Do you expect highish calcium and phosphate? So, that is reasonably consistent? What about demographic features for sarcoid? This is exactly when you see it, African Americans who are in their 30s and 40s would be perfect. No shortness of breath. No cough. Nothing like that.
Does that rule out sarcoid? No, of course not. It can be mostly extrapulmonary or it certainly can be pulmonary, but it can actually be pulmonary and the patient has no symptoms. So, chest x-rays certainly can help us because hilar lymphadenopathy would be of value. Sarcoid can also give you a tubulointerstitial disease that can present with renal dysfunction out of proportion to other abnormalities - granulomatous tubulointerstitial disease, but that is pretty uncommon. What would you do to assess for sarcoid?
Audience: 1,25,D levels? ACE level?
Dr. Bushinsky: No. Let us start with the physiology first. What physiology are you trying to capture? The physiology is - you are looking for what in sarcoid? You are trying to capture excess activation of vitamin D by granuloma leading to what? Excessive calcium absorption.
So, I would rather do a functional test, and you go down the road and get an ACE test. They do that in the case records of the New England Journal of Medicine until it is negative, but that is already the next tier, when you are reaching for a diagnosis. What you want to do is capture the physiology first, and then once you know the physiology, then your list of possibilities is much shorter. I think a 24-hour urine calcium, and if your are impatient, you might want to get a spot urine for creatinine and calcium, and you might as well get protein later, and so that sounds good. In fact, this is what I am getting at, indeed. This is sarcoidosis. What I am trying to show you is somebody who has had smoldering sarcoidosis that has not yet come to clinical attention during the hypercalcuric stage that they often are in before they develop overt hypercalcemia, and in this patient what triggered the crossing of the line to the development of hypercalcemia was the addition of the thiazide diuretic that interfered with the calciuria and then led to the hypercalcemia.
27:52

What is the treatment?
Here is the chest x-ray, showing bilateral hilar lymphadenopathy, by the way. You do not even need the ACE test, you got your answer, and then if you want to prove it with a tissue diagnosis, you send them to your pulmonologist, and they do a bronchoscopy down there and get your answer 1, 2, 3. Steroids would be the treatment of the hypercalcemia. It is sarcoidosis, absolutely, and in this case, I agree with the very first comment that you all made, which was getting rid of the hydrochlorothiazide. There are all kinds of antihypertensive choices that we can give somebody, and so certainly in this patient, we are going to avoid something that causes hypocalciuria.
28:40

Any questions?
FHH (familial hypocalciuric hypercalcemia) being in the differential. I suppose it is possible always, but you guys are very, very fixated on FHH this whole morning. It is very rare. So is sarcoid, too, I suppose. I guess it could be, but again your 24-hour urine is going to take care of you. By the way, there is one other follow-up test you can do to nail the physiology once you have the hypercalcuria. This is one of the rare circumstances where measuring the serum 1,25 D level is not a horrendous idea. You may not need to do it, because if all the footprints are there and you make a tissue diagnosis, it is not that important, but that is a situation when you are looking for excess of activation of 1,25D, it is useful sometimes in cases like this to measure it. Where it is not useful is when you are hypothesizing that there is deficiency of 1,25. Because if you are looking for a syndrome, let's say secondary hyperparathyroidism due to vitamin D deficiency, this is the most important reason for why you should not measure 1,25 routinely, in kidney patients and in vitamin D deficient patients, because what happens is when you are vitamin D deficient, let say your 25D level is 6, what does that do to your PTH? It leads to an increase in PTH through a variety of mechanisms, some more direct and some more indirect, but at the end of the day, the PTH goes up and that is the secondary hyperparathyroidism. What does that secondary hyperparathyroidism due to the 1-alpha-hydroxylase? It stimulates it, and it turns out that serum 25D levels, if you look at the units, are in ng/ml, whereas 1,25D levels are in picograms/ml, or 1000-fold higher, and so if you have lots of PTH activation even in the setting of 25D deficiency, that PTH can lead to a higher percent of activation of 25 to 1,25 and it will actually bring the 1,25 level into the low-normal range, and so if you did it, there is nothing wrong here and you would be making a big mistake. In fact, the vitamin D deficiency is the root cause of the problem. It is the exact same logic when you are talking about hypothyroidism. A T3 and T4 that is in the normal range with a TSH of 90 is very, very abnormal.
Audience: If for the question on the FHH, all you need is a PTH level. If it is high 1,25-D, what should the PTH be?
Dr. Bushinsky: It's suppressed. It's low. That's actually a great point. The calcium-sensing receptor is active and it is telling the PTH not to secrete in the setting of hypercalcemia and you have isolated the problem. That's a great point that I did not make. Thank you.
Audience: I wonder why would you treat this patient with steroids? Before hydrochlorothiazide, calcium was 10.1. The patient is completely asymptomatic.
Dr. Bushinsky: Fair enough, I would certainly give it a trial to see what happens off the thiazide, all the while I would be trying to make my diagnosis, but there are several potential things that could happen. One is that there could be chronic hypercalcuria that could ultimately lead to stone disease and to calcification in the kidney, which could potentially be a problem, and so big picture-wise, if you are going to treat somebody with hypercalcemia that becomes symptomatic and is due to this syndrome it is going to be via steroids not to mention that steroids are sometimes, not always, useful for managing the underlying sarcoidosis, but having said that I think you could certainly make a case for taking off the thiazide and see where things settle down before you went and did that, but ultimately you may end up there.
Audience: What about bone metastatic disease?
Dr. Bushinsky: The mechanism of hypercalcemia in what we have termed bone metastasis is almost always in most of these cases due to humoral hypercalcemia malignancy. They are usually PTHrP mediated. This direct metastasis from bone causing a tremendous amount of hypercalcemia certainly can happen, but it is not as common as the humoral-mediated syndrome. So, I suppose it is a possibility, but you would be able to sort through those things with some of the diagnostic tests we postulated, but if you are going to propose that it was due to a squamous cell carcinoma with overwhelming bone metastases, then that would be the mechanism and would still be PTHrP and virtually all of them.
33:49

Dr. Andress: Case 3
This was an interesting patient that came into my Osteoporosis Clinic in December. He had a six-month history of progressive arm, leg, and rib pain associated with generalized weakness, not associated with any kind of trauma. Review of systems was only positive for intermittent nausea and vomiting, decreased appetite, and weight loss. In fact, he had lost about 30 pounds over four months. Past medical history was very significant for having HIV disease for the past ten years. His current medications included those shown here as part of the HAART regimen in addition to fairly hefty doses of naproxen for his pain and a little bit of codeine.
34:57

Exam
On exam, he was very thin appearing. He came in a wheelchair. No adenopathy. On chest exam, he had definite point tenderness of the left anterior seventh and tenth ribs and the right fourth and tenth ribs. Extremities: There was mild-to-moderate tenderness of the biceps and triceps. He had symmetrically decreased muscle strength, lower extremities more so than upper extremities. Neurologically, he was nonfocal. DTRs were normal. No real abnormalities there.
Labs: Sodium 140, potassium 3.5, chloride 115, CO2 13, creatinine 1, glucose normal, albumin normal, calcium 8.5, phosphate 0.7, and magnesium 1.7 mEq/L. His UA done by the lab showed 3+ glucose, 2+ protein, and a urine pH of 5.5.
36:14

Additional Serologies
Additional serologies were obtained. We got a CPK because of the muscle tenderness, and that was normal. SGOT was normal, but his alkaline phosphatase was 332, the upper normal limit for our lab being 130. That was fractionated into a bone alkaline phosphatase of 99 with an upper normal limit of 30. Serum uric acid was 1.7. Lipase was 85. He had had a bone scan before he came to Osteoporosis Clinic. I think that is why he got into our clinic. The technetium scan showed diffuse tracer uptake in the skull, focal uptake in the right humeral head, right second, fourth, and tenth ribs, left seventh, tenth, and eleventh ribs, right proximal tibia, and the left greater trochanter.
37:14

Case 3 Labs
So, what does this patient have? I hear RTA (renal tubular acidosis) from the audience. OK, next case! Did somebody already talk about this earlier!?
Lets look at the labs. CO2 is 13 and chloride is 115, so you calculate the anion gap and it is 12. So, it is a non-gap acidosis. Urine pH is 5.5. So, it is probably not a distal RTA. Of course, we have already ruled out that the patient does not have diarrhea by history, a most common cause of non-gap acidosis. This has already put us into a ballpark of not a distal RTA. What is really revealing about this is you have got hypophosphatemia that you do not typically see very often in the osteoporosis clinic. And what is very telling about this UA is a 3+ glucose in someone who is not diabetic. He has renal glycosuria. So, you are exactly right. He has a proximal RTA or Fanconi syndrome with heavy phosphate wasting. And notice that the phosphate is lower than the magnesium, because of what I told you before, magnesium is not really processed in the proximal tubule, but it is in the thick ascending limb. Hypophosphatemia is his major problem, or at least it was the original metabolic problem that really led to what is his major problem. What's his major problem? It is on the bone scan.
You cannot make a diagnosis of osteomalacia with this scan. The only way you can actually do that is with a bone biopsy. We came very closely to doing it in him, but his whole constellation fits so well with osteomalacia that has already been reported in the literature being associated with Fanconi syndrome that we did not feel like we absolutely had to do it, but this is very typical, and I am sorry I did not have the picture to be able to show you. But this is very typical - diffuse, but usually kind of focal, uptake of hot spots where you are having increased bone formation but no mineralization, and the other biochemical feature of this is elevated bone alkaline phosphatase. What is remarkable about him is that he lived with this for six months or more and nobody really noticed that he had been acidotic all this time until he came to the Bone Clinic and just happened to see a nephrologist.
41:20

Osteomalacia with Fanconi Syndrome:
At any rate, you are right, this is tenofovir-associated Fanconi syndrome. It is probably the 20th case since a report just came out in the literature this August reviewing all the other 19 cases of tenofovir-associated Fanconi. This has all come about since 2002. It looks like it is really a potentially big problem that everybody should be paying attention to. When you review these cases, not all of them are this blatant with severe acidosis like he had.
42:10

Treatment
By the way, I will just finish up the case. We tried to treat him as an outpatient initially with 360 mEq of oral citrate a day and sodium phosphate, and he did okay. The problem was that he had so much pain and he would not get off the naproxen that he started having nausea and vomiting, just would not take anything. So, I had to bring him in the hospital and pump him up IV with bicarb and phosphate. I did an EGD. No ulcer. Just some gastritis. Put him on PPIs and metoclopramide, and his stomach did fine. All these abnormalities corrected fairly quickly. They did take him off his HAART regimen for two months while we corrected all the biochemical abnormalities and once the bone alk phos dropped from 99 down to 40 in six weeks - this was very dramatic. During the time about halfway through that he then became hypocalcemic, requiring high-dose calcitriol and calcium. His bone pain disappeared within two to three weeks, and he was up walking around. We saw him in clinic just last week. He has been off all medications for several months. All of his electrolyte abnormalities are now normalized, and he has been back on a new HAART regimen without tenofovir for a month and a half. So, it looks like by adding back on everything except the tenofovir, I think they added one other medicine here - ddI - he has done fine. It looks like it is fairly common if we are seeing this many already, and I think you need to be on the outlook.
One other clinical pearl that I picked up from this review. It appears that of the 19 cases that have been associated with tenofovir-related disease, 14 of the 19 also were on ritonavir, and ritonavir apparently causes increased accumulation of tenofovir in the proximal tubule. So, it may be that a learning point from this case, although it is not an exclusive association, is that this may not be the ideal HAART therapy to go for, at least initially.
45:00

Questions
Hypophosphatemia is often associated with rhabdomyolysis, and how low does the phosphate have to be?
Dr. Andress: I mentioned that his phosphate was 0.7, and actually that falls into the range of what we call severe hypophosphatemia and that is the point where you start seeing breakdown of muscle tissue, red cell membrane deformation, hemolytic anemia, and all these other things that go wrong.
Why his CPK was not elevated, I do not know, but I think it is a time-dependent effect, so he may not have been that low for that long, is my only guess, but he is definitely in the range where you would give him IV phosphate normally. And I debated actually when I first saw him about bringing him in right away because of that, but I did have a repeat phosphate done within a few days as an outpatient, and it has already come up to about 1.5. He responded very quickly to oral phosphate therapy. That was easy to treat interestingly. It was the acidosis that was very hard to treat, and rightly so. It is Fanconi, so whatever you put in eventually gets dumped out, and we did document that his urine pH did drop to 7 during that therapy, so again, this was further support for a diagnosis of Fanconi syndrome.
Audience: The question I believe had to do with how to give IV phosphate, short term, two to three hours versus long term, was that it? I do not know that I have played around with any kind of short-term therapy. It is usually a certain dose over eight hours, and we to repeat that. I do not know. David do you have a comment on that?
Dr. Bushinsky: When you dialyze someone, the vast majority of phosphate is removed in the first two hours because of miscible pools of phosphorus and then it comes down very slowly over the next two, four, or six hours. If you take the converse of that of giving back phosphorus, you probably achieve a lot of phosphate repletion acutely, but once you fill those miscible pools, it will go slowly, so most of us tend to give phosphorus over more prolonged periods of time, and I again push to drink milk.
Audience: In trying to evaluate hypophosphatemia, do you guys use fractional excretions of phosphorus, and if so, can you give some values as to what you think is low and high?
Dr. Bushinsky: Well, I do not use them that much, but I just did a study where I looked at this stuff, so I can at least give you an answer. Usually, hypophosphatemia that I see - most of it is in the acute setting, in which case you are not in a steady state and urinary output is not as helpful and you can usually figure out readily that it is from rhabdo or whatever it is, something acute, but when we did this study in very early kidney disease, the background fractional excretion of phosphate was between 10% and 20%, and then as people lost nephrons, it went up from there substantially. For whatever that is worth, I do not know if that will be that helpful. I think it is only going to be useful really in chronic situations like this, but you already have the clue that you knew it was renal wasting based on the glycosuria in the absence of hyperglycemia and the other clues that Dennis pointed out.
Audience: My question was when we see folks with tenofovir-related problems, should it be our universal recommendation to discontinue the drug and that clearly in this case this fellow had a lot of problems, but I find myself frequently in the position of phosphorus is a little bit low, but there is a huge viral resistance pattern that suggests that the patient has limited options.
Dr. Andress: I have really no experience dealing with these drugs. This is probably my first patient. So, I always have to go to my infectious disease colleagues. I did call them and we were going to try to do this as an outpatient, but the patient got so sick so quickly that we just decided to bring him in and make that decision. They decided to just take him off everything. I mean I do not know.
Audience: Do either of you have any experience in this? There is one case report in the literature where someone had just very minor tubular problems that progressed. They just got caught early, and that the natural history of this is that the tubular problems just developed the longer you stayed on the drug, which might be a matter of detection-wise when someone finally notes his problems?
Dr. Andress: This review article that reviewed these cases did show for tenofovir related Fanconi syndrome, the disease can strike anywhere from 1 to 26 months after being on the drug, even though little more than a half of the cases have usually occurred within the four to eight months period much like he was two years out, that is really long, so everybody is a little bit different. Some of the case reports I read had kind of mild hypophosphatemia, not really severe, and like I said, many of them did not have very bad acidosis. I think there is probably a range of disease, phenotype if you will, and how much it involves these other agents that actually cause more accumulation. The tenofovir probably is related as well.
Dr. Bushinsky: The other circumstance with the same kind of thing comes up is people with refractory CMV, and you see that sometimes post transplant, but often it is not so much now with so much good HAART therapy, but previously the CMV was with foscarnet which had some of the same problems, and cidofovir which was a CMV drug on which this tenofovir is then based, and those people who need those drugs are often at the end of the line in terms of resistance to ganciclovir and it sometimes happens in transplant patients. We had one. We gave him the foscarnet, and we knew he would get the tubular abnormalities. We ended up getting a crescentic glomerulonephritis too, but again the point is that you can see all kinds of problems in the CMV side. It comes in the same decision. Can you take them off? You may not be able to.
Dr. Andress: We quantitated his urine protein and it was less than 1 gm of protein. So, it was mainly amino acids, not albumin. He did not have a glomerular defect. It was all tubular.
Dr. Bushinsky: You have to look for Fanconi syndrome, and it is good thing the patient ran into you.
52:40

Case-4, 46-year-old female
A 46-year-old female with adult-onset diabetes of 20 years' duration, diabetic retinopathy, and diabetic gastroenteropathy. This patient was referred to us after the kidney biopsy was done. We would not have done this biopsy. So, do not yell at me for the biopsy. This is how the patient came to us. Unfortunately, a nephrologist, who did not take this course, probably ordered it. Hemodialysis was begun.
53:18

+3 Months
Plus three months, the patient was found to be hypercalcemic and was on some calcium carbonate, on some Renagel (sevelamer), and a physical exam was essentially normal.
53:31
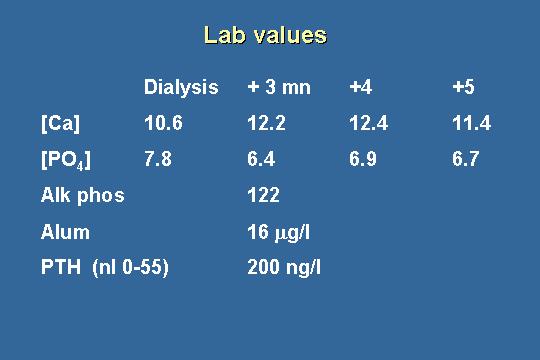
Lab values
At the time dialysis started, the calcium was 10.6 and the phosphorus was 7.8. What do you think? That is the day the patient started dialysis.
A high calcium. You would expect as the patient was developing CKD and renal failure that calcium to be on the lower side of normal, but yet the calcium is elevated. The phosphorus does not bother me very much. A lot of patients come in to dialysis a little hyperphosphatemic. We start them on binders, and about 50% of the patients do not get much below 6.4. Plus three months, when we saw the patient, the calcium was up to 12.2. By our assay, the alk phos was up a little bit. Aluminum was not interesting. The PTH was 200.
What do you think?
Audience: Primary hyperpara.
Dr. Bushinsky: So, I will sit down and we can go onto the next case. Absolutely. The patient was sent to us, and they were concerned with a high calcium in the face of initiating dialysis. Calcium stayed up. Phosphorus stayed up. Let us talk about the PTH assay, which we promised before.
55:07
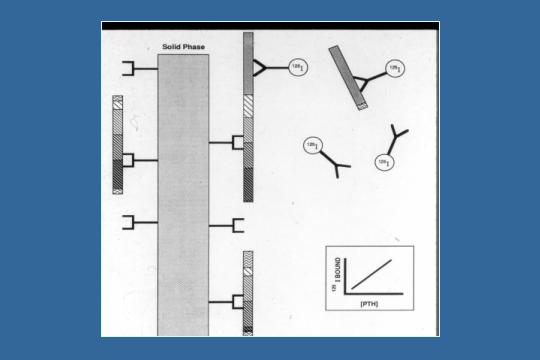
PTH Assay
.This is parathyroid hormone 1 to 84. How is the PTH assay done? You in general have a solid phase with a capture antibody. The solid phase is essentially a test tube. The capture antibody is adherent to the test tube and is sticking out. You put the serum in. You shake it up. You pour off the serum. What will remain is parathyroid hormone that is adherent to the solid phase in the area that this antibody is directed against. This is 1, this is about 34, and this is 84. If the capture antibody is directed more towards the amino-terminal, you will capture some of the amino-terminal, but if it is directed sort of at the mid terminal like this one is, you are going to correct the mid terminal. The next thing you have is the detection antibody. You shake up the test tube. You pour off the serum. You have a lot of parathyroid with a lot of fragments connected to the solid phase. You then put in a second antibody, which is the detection antibody. In this case, the detection antibody is towards the carboxy-terminal side of PTH, and this either lights up with radioactivity or immunofluorescence; there are lots of different ways to do it. You see you have detection antibody that is floating around. It's bound to this fragment of PTH, but then you pour off the soup again. You wash it a few times, and all you have left is the solid phase with the capture antibody, the PTH, and the detection antibody. First of all, we had carboxy-terminal PTH assays, which were no good at all. Then we had amino-terminal single assays, which were not much better. Now, we have the immunoradiometric assays. In this first-generation immunoradiometric assay, you see what the problem is. You have PTH 1 to 84, and the capture antibody is directed too far up the PTH molecule, so that certain fragments like 7 to 84, 10 to 84, and 3 to 84 will be detected as native PTH. People realized this, and they developed antibodies against the very tip of the amino-terminal. The newest generation of PTH assays connect by about 1 and span to about 5 right over here. They have a detection antibody that is closer to the carboxy terminal, and since it is really the first 34 amino acids in PTH that are biologically active, it picks up only biologically active PTH. It is about 50%, 54% of the old intact PTH, and it is a much cleaner assay. The world is migrating towards using those cleaner assays.
The next question you are going to ask me is, should we use the 7 to 84 since some people have shown that it is actually a suppressor. Should we use any sort of ratios? There is absolutely no data, save for one study that cannot be repeated, to show that that ratio is of any clinical use. There is the initial study. It seemed to make sense. No one else has been able to replicate that study. So we do not use any sort of ratios. We use a PTH assay that picks up very amino-terminal and carboxy terminal. We do not have firm biopsy data for any of the immunoradiometric assays with PTH, but anyway, that is how the test is done. It is much better.
Audience: Why would not you consider tertiary PTH?
Dr. Bushinsky: Good question. Because the patient has not been on dialysis very long. It takes a long time do develop tertiary PTH with absolute dysregulation of PTH secretion and autonomous secretion. She was hypercalcemic the day she started dialysis. So, while that is a good point, probably not in this patient.
60:52

+8 months
We did a parathyroid scan. A large nodule was present below the left lobe of the thyroid. Chest x-ray was normal. Mammogram was normal. We are looking for other causes of hypercalcemia.
61:06
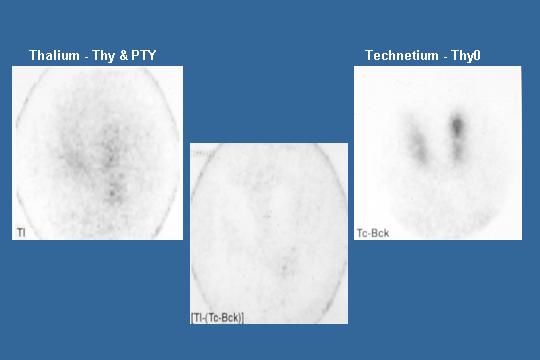
Subtraction Scans
This is a subtraction scan. In some of your hospitals, you are probably still using them. First you give the patient thallium, which picks up the thyroid and parathyroid. Then you give the patient technetium, and it picks up the thyroid and then you electronically subtract them and you see, I cannot see it very well, but there is a parathyroid gland.
61:34
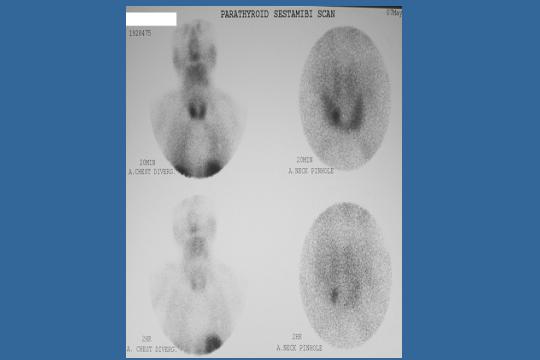
Parathyroid sestamibi scan
This is opposed to the sestamibi scan, which is on this slide, showing a different patient. You see how much better the sestamibi scan is. So, if your hospital is still doing the subtraction scan, ask them to buy sestamibi when you need to do your next scan. But clearly this gland showed up nicely.
61:57

Surgery
Surgery, she had a big adenoma. The other three glands were slightly hyperplastic. The surgeon took out 2-1/2 of the remaining three glands. Her calcium decreased to 7.9 and was 9.1 at the time of discharge. She had a little bit of hungry bone syndrome, for which we give the patients calcium and vitamin D just like you give the patients calcium and vitamin D to treat the hypocalcemia.
62:27

Hyperparathyroidism
Let us think about hyperparathyroidism. This case, primary hyperparathyroidism, autonomous secretion of PTH. In secondary hyperpara, the parathyroid gland responds to normal regulatory stimuli. It is just the parathyroid is doing exactly what it is supposed to do. It is responding to normal regulatory stimuli . In refractory secondary, you have nonsuppressible secretion of PTH after correction of the metabolic abnormalities. Remember I said every parathyroid cell has some obligate secretion of PTH and in refractory secondary, you cannot suppress that PTH. Finally, in tertiary, you have this refractory secondary with hypercalcemia.
63:24
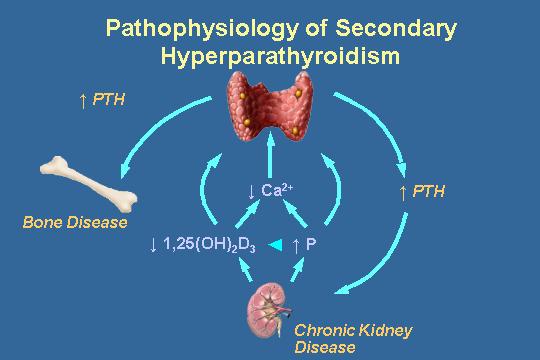
Pathophysiology of secondary hyperparathyroidism
You saw this picture before. There are a lot of iterations of this picture. It was actually started out as a pharmaceutical company slide set and it has been played with by a lot of people including myself, but just let us think about the pathophysiology. I am not sure we are going do this tomorrow.
We ingest phosphorus on a daily basis. You can't excrete the phosphorus as your GFR falls and your serum phosphorus goes up. We said phosphorus regulates the 1-alpha-hydroxylase. You have decreased 1,25 D3. The high phosphorus also decreases the 1,25 D3. This leads to hypocalcemia. The high phosphorus also leads to hypocalcemia. Hypocalcemia increases PTH secretion and causes the bone disease, but more importantly causes a phosphaturia, which self-corrects. As you are developing renal insufficiency, the high PTH causes the phosphaturia and that regulates the system. Decreased 1,25 also leads to PTH secretion. Increased phosphorus also leads to PTH secretion, but by far the best regulator is calcium.
64:54

Disordered mineral metabolism - CKD
The last slide is just to show you that this occurs very early. It does not occur in stage IV or stage V. Let us look at this collection of patients, in which the calcium, phosphorus, creatinine, and calcitriol and such were measured with each stage of CKD. Stage I: Creatinine is 1, GFR is 104, phosphorus is 3.1, PTH is 45, and calcitriol is 45. Stage II: A small bump in creatinine, GFR fell from 104 to 71, and the phosphorus is not significantly higher, but the trend is already evident. The PTH is significantly higher. This high PTH is causing the phosphaturia, which is aiming to lower the serum phosphorus. You are not absorbing as much calcium and phosphorus because the calcitriol is lower. Also, this low calcitriol allows more PTH secretion, but even in stage II, we are seeing a trend, which continues to stage III, IV, and V. The PTH continues to go up. The calcitriol continues to go down. This occurs early on. Even when the phosphorus is normal, the system, it is like looking at the temperature in this room. You do not know if the air conditioner is on or the heater is on. In this case, PTH is trying valiantly to control the phosphorus, and in spite of almost doubling the PTH, it cannot quite get the phosphorus down to where it was. Is this elevated? No. Is this higher? Yes. There is study after study showing these trends. This occurs early, and we have to intervene I think early.
Again, if you have a good surgeon and if it is primary hyperparathyroidism, there is usually a single adenoma and you take that out. In secondary, there are several things that could be going on. Sometimes, the surgeon disseminated the parathyroid cells in the surgery, and you get this systemic parathyromatosis and it is very difficult to treat, and we have been using cinacalcet in such a case, but it is a bear to treat, but that is usually the only thing. Sestamibi is pretty sensitive and specific.
I think it is up to your surgeon, and if you have a really good surgeon, you are okay. Let him do what he wants or what she wants, but I have never told a surgeon what to do. Usually, they leave gland behind. I have seen a spectacular surgeon who does it. I do not care as long as they do a lot and have good results. It is not my role to tell a surgeon how to operate.
Audience: The slide that you show is basically repetition of the Neal Bricker tradeoff hypothesis, with Slatopolsky pointing out the 60-mL figure. The problem is with the nephrologists who are not going to see those patients of stage II. Therefore, what can we do in the sense of public education or better peer education to begin to tackle this problem?.
68:34

Questions
Dr. Bushinsky: The question is we are not seeing people in stage II, but our hospital does and I am sure a lot of your hospitals do. We report GFRs now. So, the primary care provider who saw a creatinine of 1.5 who before just blew it off now sees that the GFR is 50. They send us the patient. So, I think that goes a long way. The other is education. Because we do not do it right does not mean it is right. We have to educate our primary care docs that they should send us such patients earlier. Some do. Some do not. Some of the best docs I know send them the day before dialysis.
Dr. Bushinsky: I think you are opening a can of worms. My own feeling and then I will let the rest of the people talk about it. I think we all need some vitamin D. I think there are some noncalcium phosphorus effects of vitamin D on immunologic function and lots of other things, and I think that is clearly beneficial. I think the industrial amounts of vitamin D used in an effort to lower PTH while raising calcium and phosphorus is probably not very good. You saw the results earlier of how the high phosphorus and high calcium are detrimental. My own feeling is we do not have to use those industrial amounts of 1,25 D to suppress calcium and phosphorus, but yes, a couple of mcg of Zemplar? Yes, for everybody - Let me pass it on round the table.
Dr. Andress: Going back to the stage II, I think we definitely have to educate the primary care folks to start the therapy even if they do not want to send the patient to us, for activated vitamin D. The data that are starting to come in for treatment using the KDOQI guidelines as suggested by Dr. Wolf earlier that it is not that beneficial in lowering PTH. So, probably what we are recommending is that everybody should just go on 50,000 units of ergocalciferol once a month. This is very new, and I will talk about it more tomorrow.
Dr. Andress: I was going to say in terms of the activated vitamin D story and its other effects beyond PTH, we are in an interesting pickle right now because we want to give people a treatment, namely vitamin D, that might have a variety of biological effects of which the patients are deficient very early on in kidney disease, and that at least in a stage V model has been shown to improve survival, and you have this enormous survival disadvantage in earlier stage and whether you could extend that benefit back to those people by treating them earlier is unknown, but certainly very exciting. Having said that, the big problem is that the only biological assay we currently titrate our treatments to is the PTH level. We do not know what dose will capture some of those other beneficial effects, and so in our sense, PTH management may be the least important thing that vitamin D is doing, yet it is the only thing we can link our therapy to currently. So, this is an open story. I think it is certainly better now that we have other options provided with the treatments that have less of an effect on calcium loading and phosphate loading, because certainly that needs to be avoided.
Audience: Would you advocate giving everybody vitamin D?
Dr. Andress: I say this cautiously, hopefully there is no KDOQI police in the room, but what I do in my own dialysis patients is I do give them a very small dose of IV vitamin D even if their PTH is low, and now I am biased by own research where we found that in people with a PTH less than 100, thousands of them who were treated, why they were treated I have no idea, but when you look there was a 15% of survival advantage associated with that approach. Now, those people might get ridiculed now if they did that treatment today, but at least that study when we looked at it, there was a survival advantage. So, what I do is I take that and extrapolate from that, and I will give patients 1 or 2 mcg a day even if their PTH is 40 and quite frankly anecdotally, the PTH does not go down with those baby doses. In fact, as the people get better, sometimes the PTH goes up.
I do not think you have any disagreement where we are replacing intravenous not orally. There is certainly no disagreement.
Audience: This is regarding your last slide you showed about the stage II CKD. When the patient is not having phosphaturia, we do not reinforce too much a low-phosphorus diet in stage II CKD, and I always thought that if these patients are having slightly normal high level of phosphorus, but they are having phosphaturia, we should reinforce the low-phosphorus diet so that we can prevent intact PTH going up?
Dr. Bushinsky: The question is the role of a low-phosphorus diet. Years ago, when I was in short pants, for real, Ed Slatopolsky did a very nice study where he did progressive renal ablation and phosphorus, not depletion, but concomitant reduction in oral phosphorus intake in dogs and found that they did not develop secondary or tertiary hyperparathyroidism, i.e., the phosphorus hypothesis of its importance. So, what this physician is suggesting is a low-phosphorus diet should be thought about. Absolutely. The downside of a low-phosphorus diet is that phosphorus is ubiquitous. All protein contains phosphorus. It is very difficult to get someone to truly eat a low-phosphorus diet and be adequately nutritionally sufficient. So, it is a real conundrum. We find that if we really try to protein restrict the patients, by the time they get on dialysis they are starving. We try to tell them to avoid the high-phosphorus foods, but my own feeling, and this is personal feeling, and I would ask everybody else, I would rather have them eat and not be starving by the time they get to dialysis.
Dr. Andress: I agree wholeheartedly. I think though that begs the followup question which is should we be using phosphate binders in normophosphatemic kidney disease patients and that is something that we are actually studying because I think, if you take a step back and ask what is the single trigger that leads to the development of secondary hyperparathyroidism and chronic kidney disease, nobody can tell you. Some people say well there is hypocalcemia, there is hypophosphatemia, and there is low 1, 25, and these all contribute, but what is the single thing that triggers it all. My personal bias based on some of what you already heard is that perhaps it is phosphate intake, a regular phosphate intake diet in the setting of decreased kidney mass and kidney function, that means that in order to maintain normophosphatemia, secondary mechanisms need to be recruited, the one I am interested in perhaps is FGF23, and then FGF23 excess that helps the PTH maintain normal serum levels of phosphate in turn inhibits renal 1-alpha-hydroxylase giving you calcitriol deficiency, and so that would then change if that is true, would potentially change our approach and support, maybe not phosphate restriction in the diet because it is too complicated in diabetes patients. What else can they eat? They are already on a sodium, a fat, and all these other restrictions, and if you tell them now you cannot eat protein either, there is nothing to eat but grass and cardboard, but perhaps phosphate binders might make sense.
Audience: What do you do, in terms of low intact PTH, in giving vitamin D, do not you worry about adynamic bone disease and what do you think about that?
Dr. Bushinsky: You do not want PTH to go too low - with the first-generation immunoradiometric assay, KDOQI suggests 150 to 130. That is probably a reasonable goal. If you go much lower on PTH, you run the risk of adynamic bone disease. There is a conference coming up next month in Madrid, which focuses on the fact that we are not getting bone biopsies anymore, and we really do not know what the PTHs are associated with, especially now that we have newer assays and the patients have newer vitamin D analogs. The patients are on cinacalcet, and we keep treating them and we do not even know what the bone disease is. That said, I do not worry as much about bone disease. I worry about vascular calcification because that is what is killing our patients.
Audience: Why was the alkaline phosphatase not so high? Dr. Andress: Yes, it should be high. That is why there is clinical medicine, and there are textbooks, and this is a real case.
References
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004 Aug;15(8):2208-18.
- Giachelli CM, Jono S, Shioi A, Nishizawa Y, Mori K, Morii H. Vascular calcification and inorganic phosphate Am J Kidney Dis. 2001 Oct;38(4 Suppl 1):S34-7.
| |||||||||||||||




