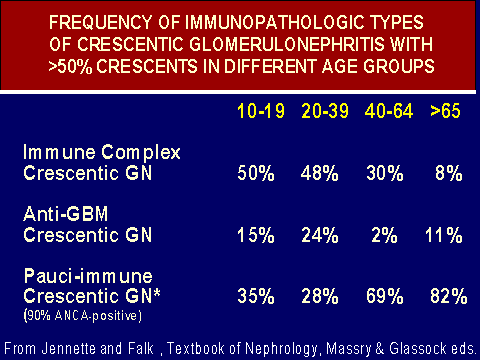
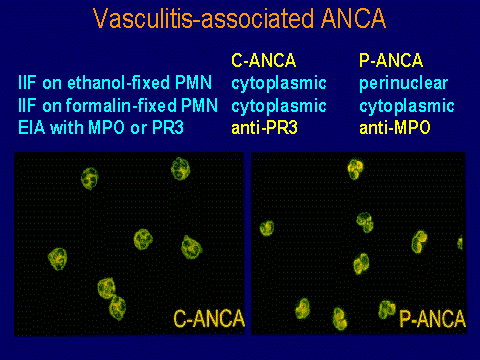
ANCA vasculitis: glomerular and extrarenal involvement
patterns
If you look at all patients with ANCA-
positive glomerulonephritis, some of them apparently have
involvement of no other tissues from all of the evidence that you
can collect. But most patients with ANCA-positive
glomerulonephritis have evidence for involvement of other viscera.
The diagnostic challenge in these patients is to categorize those
patients into some specific set if possible. This may be a
somewhat academic exercise because, as we will discuss some later,
the treatment may be very similar if not identical for all of these
categories.

So the main step is initially realizing that it is an
ANCA-positive necrotizing small-vessel vasculitis and possibly
proceeding with treatment at that point and trying to categorize
them further. If there is no systemic involvement, just ANCA
glomerulonephritis, some prefer the term renal-limited vasculitis.
If there is involvement of the respiratory tract or elsewhere with
granulomatous inflammation, then Wegener's granulomatosis is the
proper designation if there is no history of asthma. If there is
a history of asthma, then Churg-Strauss syndrome. In these
patients, the eosinophilia is usually more than 10 percent. If
there is no evidence for Wegener's granulomatosis or Churg-Strauss
syndrome, then the term that I would suggest is microscopic
polyangiitis. I believe this is preferable to microscopic
polyarteritis because many of these patients have no apparent
involvement of arteries at all. They may have pulmonary
capillaritis, they may have postcapillary venulitis in the skin,
they may have glomerulonephritis, but they may not have arteritis.
So the term microscopic polyangiitis is a better generic term.
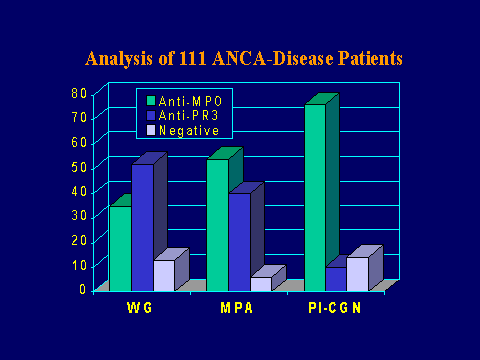
ANCA specificities
With respect to the association of the autoantibody
specificities, various cohorts have been analyzed. This is an
analysis of 111 patients from our cohort of ANCA-diseased patients.
There is a predominance of the anti-PR3 specificity in patients
with the Wegener's granulomatosis phenotype, but at least in our
experience at this point, about one-third of these patients have
anti-MPO specificity. In the microscopic polyangiitis patients,
there is about an equal frequency of anti-MPO and anti-PR3
antibodies with possibly a little bit of predominance of anti-MPO
specificity. In the patients with renal-limited disease, there is
a very striking predilection for the anti-MPO.

Chapel Hill nomenclature system
I would suggest
that the categorization of small-vessel vasculitis in patients with
renal disease can usually be accommodated by what is sometimes
referred to as the Chapel Hill nomenclature system, which
categorizes some of these major forms of small-vessel vasculitis as
Wegener's granulomatosis, Churg-Strauss syndrome, microscopic
polyangiitis, all associated with ANCA; Henoch-Schönlein
purpura,
with IgA dominant immune complexes; cryoglobulinemic vasculitis,
with circulating cryoglobulins and cryoglobulins in the tissues;
and then occasional patients who have small-vessel vasculitis just
confined to the skin. These patients may have a self-limited,
benign course, but some of these patients will ultimately be found
to have systemic involvement including the kidney.

Vasculitides not on the above list
Of course, there are other forms of small-vessel vasculitis not
accommodated
in this particular list that I've described earlier: lupus-
associated vasculitis, some forms of drug-induced vasculitis,
infection-associated vasculitis. But in all of these
circumstances, I think the initial approach should be to recognize
that your patient has a small-vessel vasculitis and then as best
you can to pursue additional diagnostic procedures, for example
immunohistology on biopsy tissue, serology, to put them into a more
specific category because that will impact substantially on the
appropriate treatment regimen.
Thank you.
References
1. Godman G, Churg J. Wegener's granulomatosis. Pathology and
review of the
literature. Arch Pathol Lab Med 1954;58:533-553.
2. Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of
systemic vasculitides:
The proposal of an international consensus conference.
Arthritis Rheum 1994;37:187-192.
3. Kallenberg CGM, Brouwer E, Weening JJ, Cohen Tervaert JW:
Anti-neutrophil
cytoplasmic antibodies: current diagnostic and pathophysiologic
potential.
Kidney Int 1994,46:1-15.
4. Jennette JC, Falk RJ: Anti-neutrophil cytoplasmic
autoantibodies: Discovery,
specificity, disease associations and pathogenic potential.
Adv Pathol Lab Med
1995; 8:363-377.
Savage COS, Harper L, Adu D. Primary systemic vasculitis.
Lancet 1997;349:553-7.
Back to Nordiska Njurdagar (Nordic Nephrology Days) Meeting
Selected Lecture List